![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
2. September 2020
Diagnose:
CHILD-Syndrom
Verlauf und Therapie
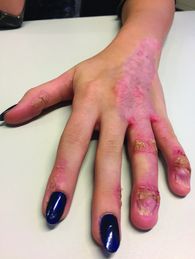
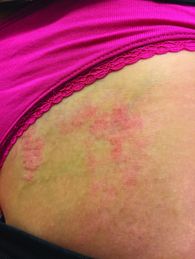
Aufgrund der richtungsweisenden Hautveränderungen in Verbindung mit Skelettanomalien sowie Hypoplasie der linken Niere stellten wir die Diagnose eines CHILD-Syndroms. Diese Diagnose konnte molekulargenetisch bestätigt werden (siehe Diskussion). Eine Pathogenese-basierte Therapie [1] wurde in modifizierter Form in Absprache mit der Universitätshautklinik Freiburg eingeleitet. Hierzu wurde eine Creme mit Simvastatin 2 % und Cholesterin 2 % in Unguentum Cordes rezeptiert. Bereits zehn Tage nach einmal täglicher Anwendung der Creme zeigte sich eine Besserung einzelner Hautareale. Nach sechs Monaten zweimal täglicher Anwendung kam es zu einer deutlichen Rückbildung (Abbildung 4), insbesondere des flächigen Areals am linken Oberschenkel (Abbildung 5).
Diskussion
Das Akronym „CHILD“ wurde von Happle eingeführt und steht für congenital hemidysplasia with ichthyosiform erythroderma and limb defects [2, 3]. Es ist ein seltenes Syndrom mit X-chromosomaler, männlich-letaler Vererbung und einer Prävalenz von 1 : 1 000 000. Zu den klinischen Symptomen zählen unterschiedlich ausgedehnte, scharf begrenzte, ichthyosiforme Areale mit wachsartiger weiß-gelblicher Schuppung. Meistens ist eine Körperhälfte mit Aussparung des Gesichtes befallen. Eine Mitbeteiligung der Gegenseite ist möglich. Diese Hautveränderungen werden begleitet von ipsilateralen Defekten der Extremitäten, die von verkürzten Metacarpalia und Finger- sowie Zehenknochen bis zum Fehlen einer ganzen Extremität reichen. Darüber hinaus sind ipsilaterale kardiovaskuläre, renale, pulmonale und ZNS-Defekte beschrieben worden [2, 3].
Ursache für das CHILD-Syndrom sind Mutationen im NSDHL-Gen, das auf dem langen Arm des X-Chromosoms (Xq28) liegt [1, 4–6]. Dieses Gen kodiert ein Schlüsselenzym der späten Cholesterinbiosynthese. Mutationen sind Diagnosequiz für männliche Betroffene in der Regel letal. Im weiblichen Geschlecht erzeugt die X-Inaktivierung ein Mosaik von Zellen, denen das Enzym fehlt, wodurch die embryonale Entwicklung gestört wird. Die Folge ist ein variables Spektrum von Anomalien. Bei unserer Patientin wurde eine heterozygote Missense- Mutation p.Ser145IIe (c.434G>T) in Exon 5 des NSDHL- Gens nachgewiesen und damit die klinische Diagnose bestätigt. Es handelt sich um eine bislang nicht beschriebene NSDHL-Mutation. Im Jahr 2011 beschrieben Paller et al. eine pathogenetisch basierte Therapie. Durch Kombination eines Statins und Cholesterin wurde eine neue topische Rezeptur entwickelt [1, 4, 5, 7]. Durch die gestörte Biosynthese kann Lanosterol nicht zu Cholesterol umgewandelt werden. Das führt zur Akkumulation von toxischen Metaboliten in Form von Cholesterinvorläufern in der Haut. Diese toxische Metaboliten verursachen Barrierefehler, wodurch die epidermale Hyperplasie entsteht. Der genaue Wirkmechanismus der Rezeptur ist noch nicht vollständig aufgeklärt. Offenbar hemmen Statine die Umwandlung von Lanosterol zu Cholesterin. Somit wird die Entstehung der toxischen Metaboliten verhindert [1, 4]. Kürzlich konnten Bajawi et al. zeigen, dass die Monotherapie mit Simvastatin 2 % einen vergleichbaren therapeutischen Effekt hat wie die bisher eingesetzte Kombinationstherapie [8].
Interessenkonflikt
Keiner.
Literatur
1 Paller A, van Steensel M, Rodriguez-Martin M et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol 2011; 131(11): 2242–8.
2 Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr 1980; 134(1): 27–33.
3 Happle R. Ptychotropism as a cutaneous feature of the CHILD syndrome. J Am Acad Dermatol 1990; 23: 763–6.
4 Elias PM, Crumine D, Paller A et al. Pathogenesis of the cutaneous phenotype in inherited disorders of cholesterol metabolism: Therapeutic implications for topical treatment of these disorders. Dermatoendocrinol 2011; 3(2): 100–6.
5 Kiritsi D, Schauer F, Wölfle U et al. Targeting epidermal lipids for treatment of Mendelian disorders of cornification. Orphanet J Rare Dis 2014; 9: 33.
6 Mi XB, Luo MX, Guo LL et al. CHILD syndrome: case report of a chinese patient and literature review of the NAD[P]H steroid dehydrogenase-like protein gene mutation. Pediatr Dermatol 2015; 32(6): e277–82.
7 Alexopoulos A, Kakourou T. CHILD syndrome: successful treatment of skin lesions with topical simvastatin/ cholesterol ointment – a case report. Pediatr Dermatol 2015; 32(4): e145–7.
8 Bajawi SM, Jafarri SA, Buraik MA et al. Pathogenesis-based therapy: cutaneous abnormalities of CHILD syndrome successfully treated with topical simvastatin monotherapy. JAAD Case Rep 2018; 4: 232–4.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft