![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
7. September 2017
Diagnose: Nicht-syndromale multiple eruptive Angiofibrome
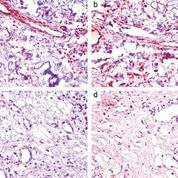
Immunhistochemie für Tuberin und Hamartin in Angiofibromen unserer Patientin (a, b) und einer Patientin mit molekulargenetisch bestätigter tuberöser Sklerose (c, d). Während Tuberin (a) und Hamartin (b) in Endothelzellen und Fibroblasten der entnommenen Angiofibrome unserer Patientin deutlich exprimiert wurden, zeigte sich ein Expressionsverlust für diese Proteine in Angiofibromen einer Patientin mit tuberöser Sklerose (c, d).
Immunhistochemie
Molekulargenetik
In der nach Aufklärung und Einverständnis der Eltern und der Patientin durchgeführten molekulargenetischen Untersuchung konnten im Vollblut der Patientin keine genomischen Mutationen in den Genen TSC1 und TSC2 nachgewiesen werden.
Diskussion
Bei unserer Patientin mit Angiofibromen sind verschiedene Tumoren/Hamartome wie multiple Neurofibrome (welche fälschlicherweise zuvor auswärts diagnostiziert wurden), eruptive Xanthome/Histiozytome oder Tricho-/Fibrofollikulome differenzialdiagnostisch zu bedenken [1, 2]. In der Regel ist hier eine sichere histologische Abgrenzung möglich; bei Angiofibromen und Tricho-/Fibrofollikulomen finden sich jedoch histologische Überlappungen mit variabler Ausprägung der hamartomatösen Bindegewebs-, Gefäß- und Haarfollikelepithelveränderungen [3]. Multiple Angiofibrome im Gesicht stellen als sogenannte Adenoma sebaceum klassische Indikatortumoren für eine tuberöse Sklerose dar [4]. In 80 % der Fälle zeigen sich diese in der frühen Kindheit [5].
Des Weiteren ist die tuberöse Sklerose gekennzeichnet durch peri- und subunguale Fibrome (Koenen-Tumoren), hypopigmentierte Maculae (Eschenlaubflecken), Bindegewebsnävi und extrakutane Manifestationen in multiplen Organen (zerebrale Tuber, renale Angiomyolipome, kardiale Rhabdomyome und Nierenzellkarzinome) [6]. Keins dieser Symptome lag bei unserer Patientin (und ihren Familienangehörigen) vor.
Ursächlich finden sich bei der tuberösen Sklerose autosomal-dominant vererbte, inaktivierende Mutationen der Tumorsuppressor-Gene TSC1 und TSC2 mit hoher Spontanmutationsrate. Eine stringente Genotyp-Phänotyp-Korrelation liegt nicht vor [7]. Die Variabilität in der klinischen Ausprägung wird durch verschiedene Mechanismen wie Zellkontext-spezifische somatische Mutationen oder epigenetisch modifizierte Genexpression beeinflusst [8, 9]. Eine konsekutive Expressionsminderung oder ein Expressionsverlust der kodierten Proteine Hamartin und Tuberin führt zur konstitutiven Aktivierung des mTOR-Signalweges mit gesteigertem zellulären Überleben und Proliferation [6] und vielgestaltiger Symptomausprägung. Auch parakrine Faktoren wie monocyte chemotactic protein 1 (MCP-1) und vascular endothelial growth factor (VEGF) spielen bei der Pathogenese der Läsionen, insbesondere auch der Angiofibrome, mit gesteigerter Gefäß- und Fibroblastenproliferation eine Rolle [6, 10]. Selten können vorwiegend faziale Angiofibrome– neben Tumoren endokriner Organe–ebenfalls im Kontext einer multiplen endokrinen Neoplasie Typ 1 (MEN1) auftreten.
Bei unserer Patientin zeigte sich bei normwertigen Serumspiegeln für Glucose, Parathormon und TSH kein Hinweis für eine endokrine Polyadenomatose im Sinne eines Hyperparathyreoidismus, eines Glukagonoms oder eines Schilddrüsenadenoms. Zusätzlich zu Trichoepitheliomen, Trichodiskomen, Fibrofollikulomen und fibrösen Nasenpapeln finden sich Angiofibrome selten auch beim Birt-Hogg-Dubé-Syndrom [2]. Diesem liegen inaktivierende Mutationen im Folliculin-Gen (FLCN) zugrunde [3, 11]. Beim Birt-Hogg-Dubé- Syndrom werden neben den kutanen Indikatorläsionen auch interne Manifestationen wie Lungenzysten und retinale Hamartome sowie selten Nierenzellkarzinome und weitere renale Tumoren, welche meist jedoch erst um die 4. Lebensdekade auftreten, beobachtet. Die klinische Ähnlichkeit mit einer überschneidenden kutanen und extrakutanen Symptomatik zwischen Birt-Hogg-Dubé-Syndrom und der tuberösen Sklerose ist wahrscheinlich darauf zurückzuführen, dass sowohl FLCN- als auch die TSC1-/2-kodierten Proteine eine regulatorische Rolle im mTOR-Signalweg spielen [12].
Im Gegensatz zu diesen „syndromalen“ Angiofibromen können multiple Angiofibrome in seltenen Fällen auch isoliert außerhalb eines syndromalen Kontextes [13] auftreten. Bislang existiert nur ein weiterer ähnlicher Fallbericht [14]. Immunhistochemisch zeigte sich in den Angiofibromen dieses Patienten eine fehlende Expression von Hamartin und Tuberin, welche sich ansonsten bei tuberöser Sklerose meist bei der pulmonalen Lymphangioleiomyomatose und renalen Angiomyolipomen, seltener bei kutanen Läsionen, findet. Möglicherweise spielen in kutanen Hamartomen eine epigenetische Regulation und ein Mosaizismus eine größere Rolle [9, 13, 15]. In unserem Fall lagen keine genomischen TSC1- oder TSC2-Mutationen in bekannten Hotspot-Regionen vor, und ein Expressionsverlust für die Proteine Hamartin und Tuberin war nicht zu verzeichnen.
Neben der Exzision oder einer ablativen Lasertherapie stellt die topische Off-Label-Anwendung des mTOR-Inhibitors Rapamycin eine innovative, allerdings sehr teure Therapiealternative bei Angiofibromen im Kontext einer tuberösen Sklerose dar [5, 16, 17]. Bei unserer Patientin haben wir einige der störenden Angiofibrome exzidiert und jährliche hautfachärztliche Kontrolluntersuchungen angeraten.
Interessenkonflikt
Keiner.
Literatur
2 Vincent A, Farley M, Chan E, James WD. Birt-Hogg-Dubé syndrome: a review of the literature and the differential diagnosis of firm facial papules. J Am Acad Dermatol 2003; 49(4): 698–705.
3 Misago N, Kimura T, Narisawa Y. Fibrofolliculoma/trichodiscoma and fibrous papule (perifollicular fibroma/angiofibroma): a revaluation of the histopathological and immunohistochemical features. J Cutan Pathol 2009; 36(9): 943–51.
4 Mentzel T, Kutzner H, Requena L, Hartmann A. [Skin tumors as marker lesions for tumor syndromes]. Pathol 2010; 31(6): 489–96.
5 Tu J, Foster RS, Bint LJ, Halbert AR. Topical rapamycin for angiofibromas in paediatric patients with tuberous sclerosis: follow up of a pilot study and promising future directions. Australas J Dermatol 2014; 55(1): 63–9.
6 Krueger DA, Northrup H, International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013; 49(4): 255–65.
7 Niida Y, Lawrence-Smith N, Banwell A et al. Analysis of both TSC1 and TSC2 for germline mutations in 126 unrelated patients with tuberous sclerosis. Hum Mutat 1999; 14(5): 412–22.
8 Jentarra GM, Rice SG, Olfers S et al. Evidence for population variation in TSC1 and TSC2 gene expression. BMC Med Genet 2011; 12: 29.
9 Niida Y, Stemmer-Rachamimov AO, Logrip M et al. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 2001; 69(3): 493–503.
10 Li S, Takeuchi F, Wang J et al. MCP-1 overexpressed in tuberous sclerosis lesions acts as a paracrine factor for tumor development. J Exp Med 2005; 202(5) :617–24.
11 Schaffer JV, Gohara MA, McNiff JM et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dubé syndrome. J Am Acad Dermatol 2005; 53(2 Suppl 1): S108–11.
12 Byrne M, Mallipeddi R, Pichert G, Whittaker S. Birt-Hogg-Dubé syndrome with a renal angiomyolipoma: further evidence of a relationship between Birt-Hogg-Dubé syndrome and tuberous sclerosis complex. Australas J Dermatol 2012; 53(2): 151–4.
13 Hunter AGW, Nezarati MM, Velsher L. Absence of signs of systemic involvement in four patients with bilateral multiple facial angiofibromas. Am J Med Genet A 2010; 152A(3): 657–64.
14 Schön MP, Ruzicka T, Wienecke R et al. Multiple eruptive angiofibromas of the trunk: case report of a new entity? Arch Dermatol 2001; 137(11): 1533–5.
15 Wataya-Kaneda M, Katayama I. Dissociate expression of tuberous sclerosis complex 1 product hamartin in a skin and pulmonary lesion of a tuberous sclerosis complex. Hum Pathol 2009; 40(3): 430–4.
16 Truchuelo T, Díaz-Ley B, Ríos L, Alcántara J, Jaén P. Facial angiofibromas treated with topical rapamycin: an excellent choice with fast response. Dermatol Online J 2012; 18(1): 15.
17 Hartford CM, Ratain MJ. Rapamycin: something old, something new, sometimes borrowed and now renewed. Clin Pharmacol Ther 2007; 82(4): 381–8.
Die Diagnosequzze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft