![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
4. Mai 2020
Diagnose:
Kutane Siderose im Rahmen einer hereditären Hämochromatose
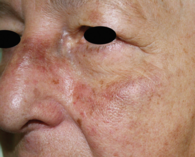
Dieses Bild aus dem Jahr 2012 zeigt im Vergleich zu Abbildung 1, dass von damals bis heute eine deutliche Progression der Läsion vorliegt.
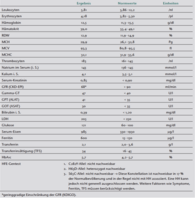
Laborergebnisse.
Ergebnis
Diskussion
Die hereditäre Hämochromatose ist eine Eisenspeicherkrankheit, die bevorzugt bei Patienten nordeuropäischer Abstammung auftritt und in 90 % der Fälle durch eine autosomal-rezessive Mutation des HFE-Gens (insbesondere C282Y und/oder H63D) verursacht wird [1, 2]. Hierdurch kommt es zu erhöhter Eisenabsorption im Duodenum und über viele Jahre zu allmählicher Eisenablagerung in Leber, Pankreas, Herz, endokrinen Drüsen, Gelenken und der Haut.
Die Erkrankung tritt meist ab dem 40. bis 60. Lebensjahr klinisch in Erscheinung. Männer sind dreimal häufiger betroffen als Frauen. Interessanterweise ist die klinische Penetration gering, etwa 30 % der C282Y-homozygoten Patienten zeigen keinerlei Symptome einer HH [2]. Etliche Faktoren wie Blutverlust (Menstruation, Schwangerschaft, Blutspende), Eisenaufnahme über die Nahrung (Protonenpumpeninhibitoren, Vitamin C) und Alkoholkonsum beeinflussen die Krankheitsausprägung [3–5]. Frühsymptome sind meist unspezifisch und können sich als Schwächegefühl, chronische Müdigkeit, Libidoverlust, Hepatomegalie, Bauch-und Gelenkschmerzen äußern [6, 7]. Hyperpigmentierung der Haut tritt bei 90 % der Hämochromatose-Patienten auf und kann ebenfalls bereits im Frühstadium sichtbar werden [7, 8]. Prädilektionsstellen sind sonnenexponierte Areale wie das Gesicht. Die Farbe variiert von Schiefergrau bis bräunlich-bronzefarben. Die typische klinische Tetrade aus Leberzirrhose, Diabetes mellitus (Bronzediabetes), Hyperpigmentierung der Haut und Kardiomyopathie wird heutzutage, aufgrund moderner Diagnostik, in weniger als 10 % der C282Y-homozygoten Patienten beobachtet [2, 9]. Um Organschäden und deren Komplikationen zu vermeiden, ist eine möglichst frühe Diagnosestellung und Behandlung der HH unerlässlich.
Auch wenn andere Differenzialdiagnosen wahrscheinlicher sind, sollte HH daher stets berücksichtigt werden, wenn Patienten die oben genannten Symptome aufweisen. Laboruntersuchungen von HH-Patienten zeigen erhöhte Werte für Serumeisen und Serum-Ferritin, die Transferrinsättigung (TFS) liegt bei über 45 %. Hiernach sollte eine genetische Untersuchung hinsichtlich HFE-Mutationen erfolgen. Eine ergänzende Leberbiopsie bei positiv getesteten Patienten sollte durchgeführt werden, wenn die Leberenzyme erhöht sind oder Ferritin > 1000 μg/l ist [6].
Auch eine Biopsie aus betroffener Haut zeigt charakteristische Veränderungen. Typischerweise können verstärkte Melaninablagerungen in der basalen Epidermis nachgewiesen werden. Da die Unterscheidung zwischen Melanin- und Eisenpigment in der HE-Färbung herausfordernd sein kann, letztere zeigen eher olivfarbene irreguläre Granulae, sollte eine ergänzende Eisenfärbung durchgeführt werden. Hier können azurfarbene Granulae um Blutgefäße und Schweißdrüsen nachgewiesen werden [7]. Eine Biopsieentnahme aus den Beinen sollte wegen möglicher Stase-bedingter Eisenablagerung vermieden werden.
Die Behandlung der HH zielt auf eine Entleerung der Eisenspeicher ab, um somit Organschäden und deren Komplikationen zu vermeiden [8]. Therapie der Wahl sind wöchentliche Aderlässe, bis Serum-Ferritin-Level von 50–100 μg/l erreicht werden. Diese werden hiernach lebenslang in individuellen Intervallen fortgeführt, um einen ausgeglichenen Eisenhaushalt zu erhalten [6]. Etliche Symptome, wie Hyperpigmentierung der Haut oder Bauchschmerzen, können sich hierunter bessern oder komplett zurückbilden. Bereits eingetretene Organschäden wie Leberzirrhose, Kardiomyopathie, Arthritis oder Diabetes sind jedoch irreversibel [6].
Bei unserer Patientin wurden die Pigmentablagerungen in allen drei Hautbiopsien zunächst als Melaninpigment interpretiert und die Diagnose Poikilodermie gestellt. Jedoch waren die Größenprogredienz über die Jahre (vergleiche Abbildung 3) sowie das atypische klinische Erscheinungsbild mit irregulären grau-braunen Hyperpigmentierungen und teils Hämatom-artigen Anteilen auffällig, sodass die histologischen Schnitte reevaluiert wurden. Eine ergänzende Eisenfärbung zeigte Eisenablagerung im Korium sämtlicher Hautproben, sodass eine weitere Diagnostik bezüglich einer Hämochromatose eingeleitet wurde. Klinisch muss differenzialdiagnostisch insbesondere an ein Angiosarkom gedacht werden. Weitere Differenzialdiagnosen sind Melasma, exogene Ochronose, Naevus Ota, medikamenteninduzierte Dyschromie (zum Beispiel Minocyclin), postinflammatorische Hyperpigmentierung, älteres Hämatom, toxisch-lichenoide Melanodermatitis [10–13]. Auch eine Kollision mehrerer Diagnosen wie Rosacea teleangiectatica und Lentigines solares wäre möglich.
Die ergänzende HFE-Gentestung zeigte eine alleinige Heterozygotie des H63D-Allels (Tabelle 1). Diese Konstellation tritt in der Regel klinisch nicht in Erscheinung, sodass Aderlässe bei diesen Patienten nicht nötig sind [14]. Interessanterweise waren Serum-Eisen und TSAT normwertig, Ferritin hingegen in mehreren Laboruntersuchungen erhöht. Die Kombination aus HFE-Gentest, Laboruntersuchung und Klinik (Hyperpigmentierung der Haut, latenter Diabetes mellitus, Hepatopathie) (Tabelle 1) beweist eindeutig das Vorliegen einer HH. Temporäre Aderlässe, als Therapie gegen die Hyperpigmentierung, wurden mit der Patientin besprochen, ein zufriedenstellendes klinisches Ansprechen ist jedoch fraglich.
Zusammenfassend handelt es sich bei der HH um eine genetisch bedingte Eisenspeichererkrankung, die bei frühzeitiger Diagnosestellung und Therapie mit einer annähernd normalen Lebenserwartung einhergeht. Durch Aderlässe sind etliche Frühsymptome reversibel. Allerdings kann die Diagnosestellung der HH, insbesondere im Frühstadium, herausfordernd sein. Es ist daher wichtig, dass Dermatologen dieses Erkrankungsbild differenzialdiagnostisch immer in Betracht ziehen, wenn Patienten atypische Hyperpigmentierungen auf sonnenexponierter Haut aufweisen.
Interessenkonflikt
Keiner.
Literatur
1 Feder JN, Gnirke A, Thomas W et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 1996; 13(4): 399–408.
2 Katsarou MS, Latsi R, Papasavva M et al. Population-based analysis of the frequency of HFE gene polymorphisms: Correlation with the susceptibility to develop hereditary hemochromatosis. Mol Med Rep 2016; 14(1): 630–6.
3 Allen KJ, Gurrin LC, Constantine CC et al. Iron-overloadrelated disease in HFE hereditary hemochromatosis. N Engl J Med 2008; 17; 358(3): 221–30.
4 Rochette J, Le Gac G, Lassoued K. Factors influencing disease phenotype and penetrance in HFE haemochromatosis. Hum Genet 2010; 128(3): 233–48.
5 Moirand R, Adams PC, Bicheler V et al. Clinical features of genetic hemochromatosis in women compared with men. Ann Intern Med 1997; 127(2): 105–10.
6 Bacon BR, Adams PC, Kowdley KV. Diagnosis and management of hemochromatosis: 2011 Practice Guideline by the American Association for the Study of Liver Diseases. Hepatology 2011; 54(1): 328–43.
7 Chevrant-Breton J, Simon M, Bourel M et al. Cutaneous manifestations of idiopathic hemochromatosis. Study of 100 cases. Arch Dermatol 1977; 113(2): 161–5.
8 Niederau C, Fischer R, Puerschel A. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology 1996; 110(4): 1107–19.
9 Durupt S, Durieu I, Nové-Josserand R. [Hereditary hemochromatosis] Rev Med Interne 2000; 21(11): 961–71.
10 Pirrone J, Broyer J, Otte N et al. Extensive hyperpigmentation of the face in a Filipina. J Dtsch Dermatol Ges 2017; 15(11): 1167–8.
11 Kreutzer K, Weber EA, Effendy I. Symmetrical hyperpigmentation of the forehead in a 37-year-old female patient from Guinea, West Africa. J Dtsch Dermatol Ges 2017; 15(10): 1042–5.
12 Wienrich BG, Bertsch HP, Schön MP et al. Dark grey hyperpigmentation on the face and neck. Diagnose: Minocycline-induced hyperpigmentation type III. J Dtsch Dermatol Ges 2010; 8(7): 544–6.
13 Fistarol SK, Itin PH. Disorders of pigmentation. J Dtsch Dermatol Ges 2010; 8(3): 187–201.
14 Porto G, Brissot P, Swinkels DW et al. EMQN best practice guidelines for the molecular genetic diagnosis of hereditary hemochromatosis (HH). Eur J Hum Genet 2016; 24(4): 479–95.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft