![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
27. September 2018
Adulter Morbus Still
Therapie und Verlauf
Aufgrund des hohen Fiebers und der erhöhten Entzündungsparameter leiten wir primär eine antibiotische Therapie mit Clarithromycin sowie eine symptomatische Therapie mit Metamizol ein. Auch unter genannter Therapie treten rezidivierende Fieberschübe bis zirka 38°C auf, die Entzündungsparameter steigen (CRP 18 mg/dl, Leukozyten 22 100/µl), es kommt zu einer Thrombozytose (410 000/µl).
Das Exanthem tritt rezidivierend meistens spät abends mit oder nach den Fieberzacken auf, ist flüchtig und im Verlauf regredient, sodass es nicht gelingt, eine Hautbiopsie zu gewinnen.
Das flüchtige Exanthem, die rezidivierenden Fieberschübe, die Arthralgien, die Leukozytose, die erhöhte BSG, die Lymphadenopathie und Splenomegalie, die erhöhten Leberfunktionsparameter, der hohe Serum-Ferritin-Wert bei negativen ANA und RF führten uns bei unserer Patientin zur Verdachtsdiagnose eines adulten Morbus Still.
Nach Ausschluss eines infektiösen und neoplastischen Geschehens leiten wir eine Kortison-Stoßtherapie mit Methylprednisolon, beginnend mit einer Dosis von 0,8 mg/ kg Körpergewicht (KG) ein. Daraufhin kommt es zu einer prompten Besserung des Allgemeinzustandes, zum Sistieren der Fieberschübe sowie zur Rückbildung der Lymphadenopathie und des makulourtikariellen Exanthems. Auch die Leukozytose, der CRP-Wert, die BSG und das Serum-Ferritin sind nach der Kortisongabe rasch regredient. Methylprednisolon wird schrittweise dosisreduziert und schlussendlich nach sechs Monaten abgesetzt. Die Patientin ist bis zum jetzigen Zeitpunkt rezidivfrei
Diskussion
Der adulte Morbus Still (adult-onset Still’s disease; AOSD) ist eine seltene Erkrankung aus dem Formenkreis der erworbenen autoinflammatorischen Syndrome des Erwachsenenalters [1]. Die Inzidenz wird in einer retrospektiven Studie in Westfrankreich mit 0,16/100 000 Einwohner angegeben [2]. Das Manifestationsalter liegt zwischen dem 16. und 50. Lebensjahr. Die Pathogenese des AOSD ist nicht gänzlich geklärt, eine hereditäre Prädisposition wird diskutiert. Es kommt zu einer Störung in der Zytokinbalance der Interleukin (IL)-1-Familie mit erhöhten IL-18 und IL-1-beta-Serumspiegeln bei bestehender Krankheitsaktivität [1].
Die Diagnose AOSD ist eine Ausschlussdiagnose und kann erst durch umfangreiche diagnostische Maßnahmen mit Hilfe diagnostischer Kriterien gestellt werden, wobei derzeit jene nach Yamaguchi et al. [3] (Tabelle 1) am häufigsten angewendet werden [1].
Die Kardinalsymptome des AOSD sind Fieber, Arthralgien oder Arthritis, Myalgien, ein charakteristisches Exanthem und Halsschmerzen. Gehäuft liegen eine Lymphadenopathie und eine Hepatosplenomegalie vor, gelegentlich kann es auch zu einer kardialen Mitbeteiligung kommen. Das klinische Spektrum ist variabel, leichte bis lebensbedrohliche Verläufe sind möglich. Bei den Laborveränderungen finden sich charakteristischerweise ein Anstieg systemischer Entzündungsparameter wie CRP und BSG, eine Hyperferritinämie, eine Leukozytose mit Neutrophilie sowie eine Transaminasenerhöhung [3–5].
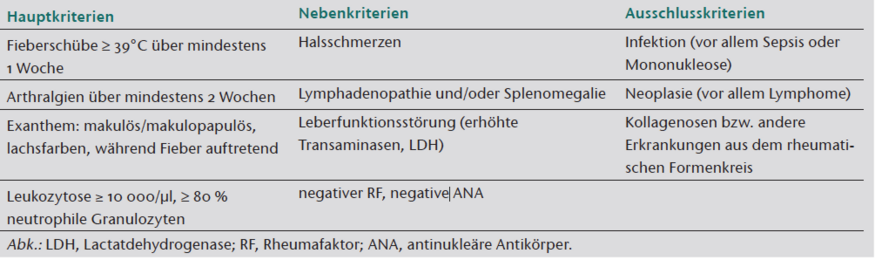
Da das Exanthem für den AOSD pathognomonisch ist und ein diagnostisches Hauptkriterium darstellt, nimmt der Dermatologe neben anderen Fachdisziplinen in der Diagnosestellung eine wichtige Rolle ein. Das Still-Exanthem ist ein meist flüchtiges hellrotes bis lachsfarbenes makulöses Exanthem, teils makulopapulös und diskret urtikariell erhaben, welches sich vorwiegend an den Extremitätenstreckseiten, am Nacken und Rumpf manifestiert. Typischerweise tritt das Exanthem mit oder unmittelbar nach einem Fieberschub auf. Es besteht ein positiver roter Dermographismus, ebenso wird ein Köbner-Phänomen beschrieben [6, 7]. Die Hautveränderungen können auch persistieren. Zur Abgrenzung von Differenzialdiagnosen ist die Gewinnung einer Probebiopsie der Haut hilfreich. In der Histologie zeigen sich in der Dermis ein schütteres perivaskuläres Infiltrat aus neutrophilen Granulozyten, Lymphozyten und Makrophagen sowie ein subepidermales Ödem mit erweiterten Gefäßen [7].
Differenzialdiagnostisch müssen neben Infektionen, Malignomen und anderen Autoimmunerkrankungen sowohl hereditäre autoinflammatorische Syndrome in Betracht gezogen werden als auch das Schnitzler-Syndrom, welches neben dem Morbus Still zu den häufigsten erworbenen autoinflammatorischen Syndromen des Erwachsenenalters zählt [1]. Das Schnitzler-Syndrom ist durch eine chronische Urtikaria, eine monoklonale Gammopathie und mindestens zwei weitere Minorkriterien wie intermittierendes Fieber, Arthralgien, Knochenschmerzen, Lymphadenopathie, Hepatosplenomegalie, erhöhte Knochendichte, erhöhte BSG und/oder Leukozytose gekennzeichnet [8].
Bisher fehlen zu den Behandlungsverfahren Daten aus randomisierten kontrollierten Studien. Die Grundlage der Therapie des AOSD sind nichtsteroidale Antirheumatika (NSAR) wie Acetylsalicylsäure und orale Glukokortikoide. Um Kortison zu sparen oder bei schlechtem Ansprechen, kann Methotrexat verordnet werden [6, 9]. Bei therapierefraktärem AOSD ist die Therapie mit einem IL-1-Rezeptor- Antagonisten anzudenken [10]. Nur bei der Hälfte der Patienten stellt sich nach Therapieeinleitung eine anhaltende Remission mit einer Normalisierung der erhöhten Ferritin- Spiegel, der Entzündungsparameter und der Leukozytose ein. Die übrigen Patienten zeigen einen chronisch progredienten oder rezidivierenden Verlauf.
Interessenkonflikt: Keiner.
Literatur
1 Lamprecht P. Adulter Morbus Still, Schnitzler-Syndrom und autoinflammatorische Syndrome im Erwachsenenalter. J Rheumatol 2009; 68: 740–6.
2 Magadur-Joly G, Billaud E, Barrier JH et al. Epidemiology of adult onset Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis 1995; 54: 587–90.
3 Yamaguchi M, Ohta A, Tsunematsu T et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol 1992; 19: 424–30.
4 Dechant C, Krüger K. Adulter Morbus Still. Dtsch Med Wochenschr 2011; 136: 1669–73.
5 Braun-Falco M, Ruzicka T. Skin manifestations in autoinflammatory syndromes. J Dtsch Dermatol Ges 2011; 9: 232–45.
6 Baerlecken NT, Schmidt RE. Adulter Morbus Still, Fieber, Diagnose und Therapie. Z Rheumatol 2012; 71: 174–80.
7 Sunderkötter C, Frieling U, Nashan D, Metze D. Morbus Still im Erwachsenenalter (adult onset Still´s disease): Erkrankung mit einem charakteristischen Exanthem. Hautarzt 1998; 49: 920–4.
8 Eiling E, Schröder JO, Gross WL et al. The Schnitzler syndrome: chronic urticaria and monoclonal gammopathy – an autoinflammatory syndrome? J Dtsch Dermatol Ges 2008; 6: 626–31.
9 Efthimiou P, Paik PK, Bielory L. Diagnosis and management of adult onset Still’s disease. Ann Rheum Dis 2006; 65: 564–72.
10 Kötter I, Wacker A, Koch S et al. Anakinra in patients with treatment resistant adult-onset Still’s disease: four case reports with serial cytokine measurements and a review of the literature. Semin Arthritis Rheum 2007; 37: 189–97.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft