![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
29. August 2017
Lichen ruber inversus
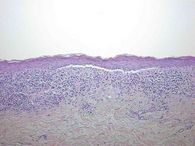
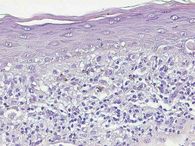
Histologischer Befund
Strukturarme Epidermis mit ausgeprägtem, teils keilförmigem Stratum granulosum und kompakter hyperorthokeratotischer Hornschicht. In basalen Epidermisanteilen vakuoläre Degeneration und reichlich nekrotische Keratinozyten. Fokal subepidermale Spaltbildung. Ein dichtes bandförmiges, lichenoides, vorwiegend lymphozytäres Entzündungsinfiltrat grenzt an die Epidermis an. Deutliche Pigmentinkontinenz (Abbildungen 2, 3).
Diskussion
Der Lichen ruber ist eine chronisch entzündliche, in der Regel selbstlimitierende Dermatose mit unklarer Ätiologie, die an Haut, Schleimhaut und seltener an Adnexen auftritt. Die molekulare Pathogenese ist noch nicht vollständig geklärt. Eine Autoimmunreaktion wird diskutiert, bei der CD8+-T-Lymphozyten Keratinozyten angreifen und somit deren Apoptose induzieren [1]. Der Autoimmunprozess kann durch multiple Faktoren induziert und beeinflusst werden, z. B. durch virale oder bakterielle Antigene, Metallionen, Medikamente oder physikalische Faktoren. Welche Rolle einzelne Triggerfaktoren spielen, bleibt allerdings noch unklar, es fehlen die eindeutigen Nachweise, insbesondere auch über die Rolle der
Hepatitisviren.
Zusammenhänge zwischen einer Hepatitis-C-Infektion und dem Lichen ruber wurden berichtet [1, 2]. Die Prävalenz des Lichen ruber planus liegt bei ca. 0,5 % (0,14 %–0,80 %); dieser ist somit eine relativ häufige Hautkrankheit [3]. Der Erkrankungsgipfel liegt zwischen dem 30. und 60. Lebensjahr. Familiäre Formen sind bekannt, aber selten, sie manifestieren sich in der Kindheit oder in der frühen Adoleszenz [1].
Der Lichen ruber tritt in vielfältigen klinischen Varianten auf, teilweise mit sehr unterschiedlicher Morphe und Verteilung. Dadurch ergibt sich ein breites Spektrum an Differenzialdiagnosen. Die klassische Form des Lichen ruber präsentiert sich mit meist intensiv juckenden, rötlichen und rötlich-violetten, polygonalen flachen Papeln [1, 4]. Diese können sich „flechtenartig“ ausbreiten und zu Plaques konfluieren. Auf der Oberfläche der aggregierten Papeln ist insbesondere mit dem Dermatoskop das pathognomonische weißliche Netzwerk zu sehen, die sogenannten Wickham-Streifen, die durch Verdickung der keratohyalinhaltigen Zellschichten des Stratum granulosum entstehen. Prädilektionsstellen sind die Beugeseiten der mittleren und distalen Extremitätengelenke, die Unterschenkelstreckseiten sowie die Sakralregion. Nicht selten, manchmal sogar ausschließlich, ist die orale oder genitale Schleimhaut befallen [2].
In unserem Fall war die Verteilung in den Intertrigines typisch für einen Lichen ruber inversus, was eine sehr seltene Form der Erkrankung darstellt; es gibt bisher nur wenige publizierte Fälle [1, 5]. Im Unterschied zum klassischen Lichen ruber verursachen die Hautveränderungen keinen oder nur milden Juckreiz, wie es auch bei unserer Patientin der Fall war. Aufgrund der Morphe und Verteilung vermuteten wir zunächst einen Lichen sclerosus et atrophicans. Weitere Differenzialdiagnosen, wenn auch weniger wahrscheinlich, waren eine persistierende Pityriasis versicolor (schon mehrere Monate mit Antimykotika behandelt), ein Erythrasma, eine inverse Psoriasis, ein Lupus erythematodes sowie ein Morbus Dowling-Degos.
Trotz der großen klinischen Vielfalt ist der histologische Befund in der Regel eindeutig und diagnoseführend. Typische Merkmale sind die Zerstörung basaler Keratinozyten und die lichenoide Interfacedermatitis. Somit ließen sich die oben aufgeführten Differenzialdiagnosen ausschließen und ein Lichen ruber bestätigen. Die ebenfalls typische sägezahnartige Akanthose fehlte in unserem Fall, sodass man histologisch auch ein Erythema dyschromicum perstans (ashy dermatosis) diskutieren kann. Klinisch fehlten jedoch die stammbetonte Verteilung und die bei Erythema dyschromicum perstans zu erwartenden gräulichen Aspekte der Läsionen. Aus unserer Sicht war die nicht ausgeprägte Akanthose auf die vorliegende Hautatrophie zurückzuführen. Am schwierigsten ist die Abgrenzung zu einer lichenoiden Arzneimittelreaktion, die aber durch Plasmazellen, eosinophile Granulozyten und weniger dichtes lymphozytäres Infiltrat charakterisiert ist [6]. In unserem Fall fehlten diese Merkmale sowie eine entsprechende Anamnese.
Interessenkonflikt
Keiner.
Literatur
1 Wagner G, Rose C, Sachse MM. Der Lichen ruber planus und seine klinisch-morphologischen Varianten. J Dtsch Dermatol Ges 2013; 11: 309–21.
2 Vogt. Papulöse und lichenoide Erkrankungen. In: Braun-Falco O, Plewig G, Wolff HH et al. (Hrsg.): Dermatologie und Venerologie. Berlin, Heidelberg, New York: Springer Verlag, 2012: 649–64.
3 Wolf R, Ruzicka T, Rupec RA. Pleomorphismus des Lichen ruber – klinische Variationsbreite, Pathogenese und Therapie. Akt Dermatol 2010; 36: 180–5.
4 Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol 1991; 25: 593–619.
5 Ohshima N, Shirai A, Saito I et al. Lichen planus pigmentosus inversus occurring extensively in multiple intertriginous areas. J Dermatol 2012; 39: 412–4.
6 El Shabrawi-Caelen L, Soyer HP. Entzündliche Dermatosen der dermoepidermalen Junktion (Interface-Dermatitis). In: Kerl H, Garbe C, Cerroni L et al (Hrsg.): Histopathologie der Haut. Berlin, Heidelberg, New York: Springer Verlag, 2003: 117–43.
Die Diagnosequzze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft