![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
22. November 2017
Acrodermatitis enteropathica
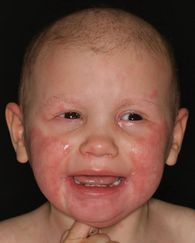
Paraklinische Befunde
Der Serumzinkspiegel war initial mit 12,0 μ mol/l im Normbereich (11,5–15,3 μ mol/l), alkalische Phosphatase im Verlauf mit 104 U/l leicht erniedrigt (110–590 U/l), geringe Anämie (Hämoglobin 11,3 g/dl, Erythrozyten 3,92/pl) sowie Nachweis von Enterobacter spp. und Pseudomonas spp. auf erosiven Plaques; kein Wachstum von Pilzen. Die im weiteren klinischen Verlauf eingeleitete molekulargenetische Untersuchung ergab eine Compound-Heterozygotie für c.283C>T (p.R95C) und c.1109dupG (p.A371fs*47) im SLC39A4 -Gen.
Diagnose, Therapie und Verlauf
Aufgrund der Klinik bestand trotz normwertiger Serumzinkspiegel der Verdacht auf eine Zinkmangeldermatitis, sodass eine orale Substitution mit Zinkorotat (Tagesdosis von 2 mg/kg KG) eingeleitet wurde, die innerhalb von drei Wochen zu einer raschen Befundbesserung (Abbildung 2 ) führte, jedoch ebenso zu schnellen Rezidiven bei Auslassversuchen. Die eingeleitete molekulardiagnostische Untersuchung bestätigte bei unserer Patientin die Diagnose einer Acrodermatitis enteropathica. Das Mädchen ist unter einer dauerhaften systemischen Zinksubstitution
symptomfrei und zeigte eine unauffällige körperliche Entwicklung.
Diskussion
Zink gehört zur Gruppe der essentiellen Elemente und
spielt in ionisierter Form eine wichtige Rolle in der Biosyntheseleistung
der Zelle, zudem ist Zink ein Kofaktor von
über 100 Enzymen und ist in mehrere wichtige Stoffwechselwege
involviert. Zink muss extern mit der Nahrung zugeführt
werden und ist hierbei eng an den Proteingehalt der
Nahrungsmittel gebunden. Die Tagesdosis bei Erwachsenen
liegt bei 16 mg, bei Schwangeren und Stillenden 25 mg und
bei Säuglingen 5 mg.
Auch bei dem klinischen Bild einer Zinkmangeldermatitis
kann der Serumspiegel normwertig sein. Dieser paradoxe
Befund ist zum Teil durch die Regulation des Zinkplasmaspiegels,
der nur 0,1 % des gesamten Zink widerspiegelt, bedingt.
In der Regel sollte das klinische Bild wegweisend sein,
jedoch gleichzeitig mit paraklinischen Befunden im Verlauf
untermauert werden [ 1 ] .
Zinkmangel im Säuglings- und Kindesalter kann primär
durch eine genetisch bedingte Störung der Zinkresorption,
der Acrodermatitis enteropathica, oder sekundär durch
mangelnde exogene Zufuhr, die erworbene Zinkmangeldermatitis,
entstehen. Der häufi g gebrauchte Begriff der Acrodermatitis
enteropathica sollte ausschließlich den genetisch
bedingten Fällen vorbehalten bleiben. Abgesehen vom Manifestationsalter
sind die erworbene Zinkmangeldermatitis und
die genetisch bedingte Acrodermatitis enteropathica klinisch
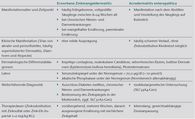
kaum zu unterscheiden (Tabelle 1 ). Bei der Acrodermatitis
enteropathica handelt es sich um eine unbehandelt tödlich
verlaufende Erkrankung.
Die erworbene Zinkmangeldermatitis tritt typischerweise
bei voll gestillten Säuglingen zwischen der 8. und 24. Lebenswoche
auf, besonders betroffen sind Frühgeborene [ 2 ] .
Relevante Zinkübertragungen von der Mutter auf das ungeborene
Kind fi nden besonders im 3. Trimenon statt, so dass
Frühgeborene bereits mit niedrigeren Zinkspiegeln zur Welt
kommen. In den ersten Monaten verzeichnen viele Frühgeborenen
nach den überstandenen Anpassungsstörungen der
ersten Wochen starke Wachstumsschübe mit erhöhtem Zinkbedarf
und gelangen damit in eine negative Zinkbilanz [ 3 ] .
Selten kann auch bei vollgestillten Neugeborenen bedingt
durch einen verminderten Zinkgehalt der Muttermilch bei
gleichzeitig normwertigem mütterlichem Serumzinkspiegel ein Zinkmangel auftreten. Ursächlich konnte in diesen Fällen eine maternale Heterozygotie für eine Mutation im
SLC30A2 -Gen nachgewiesen werden [ 4 ] . Ein sekundärer
Zinkmangel kann auch Folge von Mangelernährung, Diabetes
mellitus, chronischer Nieren- oder Darmerkrankung
sowie Mukoviszidose sein [ 5 ] .
Diese Situation ist abzugrenzen von der genetisch bedingten
Acrodermatitis enteropathica, die durch eine gestörte
Zinkaufnahme im Duodenum und Jejunum aufgrund einer
Mutation eines Zink-Transportproteins bedingt ist [ 6 ] . Die
genetisch bedingte Acrodermatitis enteropathica ist eine
durch Mutationen im SCL39A4-Gen bedingte und auf dem
Chromosom 8q24.3 lokalisierte autosomal-rezessive vererbte
Erkrankung [ 7 ] . SLCA39A4 kodiert den Zink-Transporter
ZIP4, der an Enterozyten exprimiert wird. Mittlerweile sind
über 30 verschiedene Mutationen im SCL39A4 Gen beschrieben.
Die Acrodermatitis enteropathica manifestiert sich in
der Regel erst nach dem Abstillen bzw. nach Umstellung des
Säuglings auf Kuhmilch, weil diese im Vergleich zur Muttermilch
weniger exogenes Zink-Eisen-Transportprotein enthält.
Insgesamt haben Patienten mit ausgeprägtem Zinkmangel
charakteristische kutane, gastrointestinale, ophthalmologische
und psychische Symptome. Zu der typischen dermatologischen
Manifestation gehören sowohl eine akral als auch
periorifi ziell ausgeprägte, häufi g superinfi zierte Dermatitis
mit akraler Bläschenbildung und mit charakteristischerweise
scharfer Begrenzung der entzündlichen Plaques, eine Alopezie,
eine Paronychie und eine Onychodystrophie.
Differenzialdiagnostisch sollte in Abhängigkeit von der
klinischen Ausprägung und dem Erstmanifestationsalter an
eine Impetigo contagiosa, bullöse Dermatosen, seborrhoisches
Ekzem, mukokutane Candidose sowie an andere Mangelzustände
wie Biotinmangel, Mangel an essentiellen Fettsäuren
und Organoazidopathie gedacht werden. Auch die
infantile Psoriasis und die Windeldermatitis sind potenzielle
Differenzialdiagnosen.
Die gastrointestinale Symptomatik umfasst schwere Diarrhoen
und es kann bei den Patienten bei längerem Verlauf
zu Gedeihstörung, Anämie, Wachstumsrückstand und verminderter
Leistungsfähigkeit kommen.
Die Therapie der Wahl ist die sofortige Zinksubstitution
mit Zinksulfat oder Zink-DL-Aspartat 1–2 mg/kg KG, was
zu einer oft raschen Befundbesserung innerhalb von wenigen
Tagen führt [ 8 ] . Eine kurative Behandlung der Acrodermatitis
enteropathica ist nicht möglich. Im Therapieverlauf sollte
nicht nur der Zinkspiegel im Serum kontrolliert werden, sondern
auch die Aktivität der alkalischen Phosphatase im Serum,
deren Anstieg den Therapieerfolg anzeigt. Die alkalische
Phosphatase ist ein typisches zinkabhängiges Enzym, deren
Aktivität bei Zinkmangel erniedrigt ist.
Fazit
Das Zusammentreffen charakteristischer akraler und periorifi
zieller Hautsymptome, Diarrhoe sowie eine potenzielle
Malnutrition nach dem Abstillen muss immer an einen genetisch
bedingten Zinkmangel denken lassen. Im Falle eines
erniedrigten Zinkspiegels und/oder einer verminderten
Aktivität der alkalischen Phosphatase im Serum sowie einer
kompletten Rückbildung unter Zinksubstitution und einem
Rezidiv der klinischen Symptomatik nach Absetzen der
Zinksubstitution sollte eine genetische Diagnostik erfolgen.
Danksagung
Wir danken Herrn Prof. Kohlhase, Praxis für Humangenetik,
Freiburg, für die molekulargenetische Untersuchung.
Interessenkonflikt
Keiner.
Literatur
1 Maverakis E , Fung MA , Lynch PI et al. Acrodermatitis enteropathica
and an overview of zinc metabolism . J Am Acad Dermatol
2007 ; 1 : 116 – 24 .
2 Benedix F , Hermann U , Brod G et al. Transiente Zinkmangeldermatitis
des Frühgeborenen . Hautarzt 2008 ; 59 : 563 – 6 .
3 Coelho S , Fernandes B , Rodrigues F et al. Transient zinc deficiency
in a breast fed, premature infant . Eur J Dermatol 2006 ;
16 : 193 – 5 .
4 Chowanadisai W , Loennerdal B , Kelleher SL . Identification of
a Mutation in SLC30A2 (ZnT-2) in Woman with Low Milk Zinc
Concentration That Results in Transient Neonatal Zinc Deficiency
. J Biol Chem 2006 ; 281 : 39699 – 707 .
5 Zedek D , Morrell DS , Graham M et al. Acrodermatitis enteropathica
– like eruption and failure to thrive as presenting signs
of cystic fibrosis . J Am Acad Dermatol 2008 ; 2 : 5 – 8 .
6 Jung AG , Mathony UA , Behre B et al. Acrodermatitis enteropathica:
an uncommon differential diagnosis in childhood –
first description of a new sequence variant . J Dtsch Dermatol
Ges 2011 ; 9 ( 12 ): 999 – 1002 .
7 Wang K , Pugh EW , Griffen S et al. Homozygosity mapping
places the acrodermatitis enteropathica gene on chromosomal
region 8q24.3 . Am J Hum Genet 2001 ; 68 : 1055 – 60 .
8 Poblete-Gutiérrez P , Wiederholt T , Frank J . Hereditäre Stoffwechselerkrankungen
mit kutaner Manifestation . Hautarzt
2004 ; 5 : 952 – 9 .
Die Diagnosequzze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft