![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
27. August 2019
Hereditäre Hypotrichose Typ Marie Unna
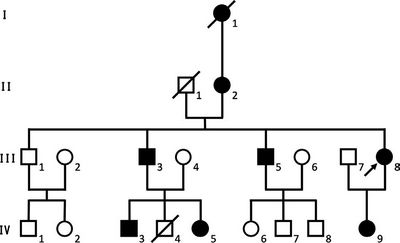
Stammbaum der untersuchten Familie. Männer sind mit Vierecken dargestellt, Frauen mit Kreisen. Alle betroffenen Familienmitglieder sind mit schwarz gefüllten Symbolen
dargestellt, verstorbene Individuen mit einem Diagonalstrich. Die Index-Patientin (Individuum III-8) ist mit einem Pfeil gekennzeichnet.
Diskussion
Die Hypotrichose Typ Marie Unna (MUHH; OMIM 146550) ist eine seltene, autosomal-dominant vererbte, non-syndromale Genotrichose. Die Erstbeschreibung der Erkrankung erfolgte im Jahr 1925 durch die deutsche Dermatologin Dr. Marie Unna in einer sieben Generationen umfassenden norddeutschen Familie [1]. Seitdem wurden mehr als 25 weitere Patienten und Familien beschrieben [2–7]. Die exakte Prävalenz und Inzidenz der MUHH ist nicht bekannt.
Der Erkrankung liegt ein genetisch bedingter Strukturdefekt des Haarschaftes zu Grunde. Dieser wird klinisch durch eine in ihrer Ausprägung variable Hypotrichose reflektiert (Abbildung 1a–d), die sich in verschiedenen Lebensabschnitten manifestiert und mit voller Penetranz in betroffenen Familien segregiert (Abbildung 2). Bei Geburt imponieren spärliche oder nicht ausgeprägte Augenbrauen und Wimpern, wie auch bei den hier beschriebenen Patientinnen (Abbildung 1a, c). Das Haar am Capillitium wächst nur sehr langsam und ist in der Regel dunkelbraun bis schwarz [4]. In der frühen Kindheit zeigt es sich zumeist spröde und brüchig und weist einen drahtig von der Kopfhaut abstehenden, mitunter aber auch gewellten oder lockigen Aspekt auf [3–7]. In der Pubertät kommt es am behaarten Kopf zu einem progredienten Haarausfall ohne klinische oder histopathologische Entzündungszeichen. Die übrige Körperbehaarung, insbesondere der Axillar- und Schambereich, ist für gewöhnlich spärlich [4, 7, 8]. Die weiteren ektodermalen Strukturen sind üblicherweise normal ausgebildet, auch wenn in Einzelfällen eine moderate Hyperhidrose und Vergrößerung des Diastema mediale beschrieben worden ist [4, 7].
Falls der klinische Verdacht auf eine MUHH molekulargenetisch nicht bestätigt werden kann, sollte differenzialdiagnostisch an weitere autosomal-dominant vererbte non-syndromale Formen der Hypotrichose gedacht werden (Tabelle 1) [3].
Biochemische Laboruntersuchungen zeigten bei den Betroffenen Normwerte für Blutbild, Eisen, Dehydroepiandrosteronsulfat und follikelstimulierendes Hormon [6, 7]. Darüber hinaus wurden in diagnostischer Hinsicht von verschiedenen Autoren licht- und rasterelektronenmiskroskopische Untersuchungen an Patienten mit MUHH durchgeführt. Zwei Autorengruppen berichteten über licht-mikroskopisch vergrößerte Diameter der Haupthaare sowie fokale Trichoschisis und longitudinale Rillen in der Elektronenmikroskopie [4, 6, 7]. Es ist jedoch unklar, ob diese Befunde pathognomonisch für die MUHH sind, insbesondere da in der Regel nicht alle Haare betroffen sind und das Ausmaß der Veränderungen schwankt [4, 6]. Dies ist in Übereinstimmung mit der lichtmikroskopischen Analyse der Haare unserer Patientinnen, die unauffällig war.
Im Jahr 2009 berichteten Wen et al. erstmals über Mutationen im U2HR-Gen auf Chromosom 8p21-p22 als Ursache der MUHH. U2HR kodiert für das gleichnamige Peptid U2HR, das aus 34 Aminosäuren besteht. U2HR bildet einen von vier offenen Leserahmen (U1HR–U4HR), die in der nicht-translatierten 5’-Region des humanen hairless (HR)-Gens lokalisiert sind. Es übt unter physiologischen Bedingungen eine regulatorische Inhibition auf das nachgeschaltete HR-Gen aus, die insbesondere während des Anagens, also der Wachstumsphase des Haars, von essenzieller Bedeutung ist [5, 7, 8]. HR wirkt als transkriptioneller Ko-Repressor für verschiedene nukleäre Rezeptoren, wie beispielsweise den Thyroidhormon- und Vitamin-D-Rezeptor. Mutationen in U2HR führen durch den Verlust der regulatorischen Inhibition zu einer HR-Überexpression, was für das Krankheitsbild der MUHH verantwortlich gemacht wird [5, 7–9]. Bei der Index-Patientin und ihrer Tochter fanden wir mittels Sanger-Sequenzierung eine bislang nicht beschriebene heterozygote C>A-Transversion, die an Aminosäurenposition 19 des U2HR-Proteins zur Nonsense-Mutation c.57C>A;p. C19* führt. Somit konnte die klinische Diagnose einer MUHH molekulargenetisch in der Familie bestätigt werden.
Derzeit ist keine Therapie für die MUHH bekannt. Behandlungsversuche mit Glukokortikosteroiden oder Minoxidil erwiesen sich als erfolglos [6], so dass den Betroffenen lediglich das Tragen einer Perücke bleibt. Die Entschlüsselung der genetischen Basis seltener Genotrichosen hat in den zurückliegenden Jahren zu einem besseren Verständnis häufiger Haarverlusterkrankungen beigetragen, da die mutierten Gene oftmals für Proteine kodieren, die eine Schlüsselposition in molekularen Signalrouten haben und somit unter translationalen Gesichtspunkten für die Entwicklung zielgerichteter Therapiestrategien von großer Bedeutung sind [9, 10].
Danksagung
Wir danken der Familie für Ihr Interesse und ihre Mitwirkung an dieser Untersuchung. Matthias Andreas Hermasch wurde durch ein von der Else-Kröner-Fresenius-Stiftung finanziertes Stipendium des Göttinger Promotionskollegs für Medizinstudierende (2017_Promotionskolleg.04) und das Dr.-Kurt-und-Eva-Herrmann-Stipendium der Alfred-Mar-chionini-Stiftung unterstützt. Regina C. Betz ist ein Mitglied des von der Deutschen Forschungsgemeinschaft (DFG) geförderten Exzellenzclusters Immunosensation.
Interessenkonflikt
Keiner.
Literatur
1 Unna M. Über Hypotrichosis congenita hereditaria. Dermatologische Wochenschrift 1925; 81: 1167–78.
2 Yang J, Liang Y, Zeng K et al. Marie Unna hereditary hypotrichosis: a recurrent c.74C>T mutation in the U2HR gene literature review. Int J Dermatol 2014; 53: 206–9.
3 Betz RC. [Alopecia and hypotrichosis in childhood: clinical features and diagnosis]. Hautarzt 2014; 65: 520–6.
4 Roberts JL, Whiting DA, Henry D et al. Marie Unna congenital hypotrichosis: clinical description, histopathology, scanning electron microscopy of a previously unreported large pedigree. J Investig Dermatol Symp Proc 1999; 4: 261–7.
5 Zhou C, Zang D, Ma X et al. Identification of a novel U2HR mutation c.14C>T in a Chinese patient with Marie Unna hereditary hypotrichosis. Eur J Dermatol 2012; 22: 34–5.
6 Podjasek JO, Hand JL. Marie-Unna hereditary hypotrichosis:case report and review of the literature. Pediatr Dermatol 2011; 28: 202–4.
7 Mansur AT, Elcioglu NH, Redler S et al. Marie Unna hereditary hypotrichosis: a Turkish family with loss of eyebrows and a U2HR mutation. Am J Med Genet A 2010; 152A: 2628–33.
8 Wen Y, Liu Y, Xu Y et al. Loss-of-function mutations of an inhibitory upstream ORF in the human hairless transcript cause Marie
Unna hereditary hypotrichosis. Nat Genet 2009; 41: 228–33.
9 Has C, Sitaru C. Molecular dermatology comes of age. Methods Mol Biol 2013; 961: 1–16.
10 Frank J. Hereditäre Hauterkrankungen – Klinisch und genetisch heterogen. J Dtsch Dermatol Ges 2017; 15: 881–2.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft