![Logo: Junge Dermatologen im Berufsverband der deutschen Dermatologen - zur Startseite [ALT+1]](/typo3conf/ext/judermconfig/Resources/Public/Images/Branding/JuDermLogo.png?v=2)
Diagnose
5. Mai 2020
Diagnose:
Paraneoplastische vesikulobullöse Dermatomyositis
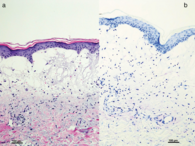
Hautbiopsie: Subepidermale Blasenbildung mit einem massiv ödematös aufgelockerten Papillarkörper und Erythrozytenextravasaten. Fehlen des Stratum basale. Im oberen Korium schütteres diffuses Infiltrat aus Lymphozyten und einzelnen Eosinophilen, Hämatoxylin- Eosin-Färbung, Originalvergrößerung x 100 (a). In der Toluidinfärbungbestätigen sich Muzin- Ablagerungen, Originalvergrößerung x 100 (b).
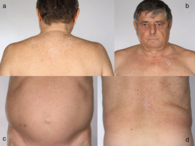
Befund nach sechs Monaten. Rückläufige Hautbefunde mit abgeblassten Erythemen, verheilten Ulzerationen und residueller Poikilodermie (a–d).
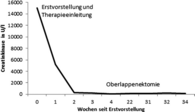
Verlauf der Creatinkinase.
Histologie
In der histologischen Untersuchung (Abbildung 2a) zeigte sich eine subepidermale Blasenbildung mit einem massiv ödematös aufgelockerten Papillarkörper und Erythrozytenextravasaten. Das Stratum basale der Epidermis fehlte und im oberen Korium lag ein schütteres diffuses Infiltrat aus Lymphozyten und einzelnen Eosinophilen. Außerdem zeigten sich Ablagerungen von Muzin, die mittels Toluidinfärbung bestätigt werden konnten (Abbildung 2b).
Labordiagnostik
Die serologische Untersuchung zeigte eine stark erhöhte Creatinkinase (CK), initial bis 15.000 U/l (Normwert < 170 U/l) und erhöhte Werte für LDH, GOT und GPT.
Im serologischen Autoimmunscreening auf antinukleäre Antikörper (ANA) mittels indirekter Immunfluoreszenz an HEp2-Zellen und nachfolgender Differenzierung mittels ELISA ergaben sich ein negativer ANA-Screen und negative Werte für Anti-Mi-2 Antikörper und Anti Jo-1 Antikörper. In der direkten Immunfluoreszenz waren keine Ablagerungen nachweisbar.
Wir stellten in Zusammenschau von Klinik, Histologie und der auffälligen CK-Erhöhung die Diagnose einer bullösen Dermatomyositis (DM).
Aufgrund der Schwere des klinischen Erscheinungsbildes, veranlassten wir bei Verdacht auf ein paraneoplastisches Geschehen zum Ausschluss eines Malignoms zunächst eine radiologische Untersuchung der Lunge. Diese wurde angesichts eines malignitätssuspekten Befundes weiter mittels PET-CT und Bronchoskopie abgeklärt. Der pathologische Befund ergab ein Plattenepithelkarzinom G3 (nicht-kleinzelliges Bronchialkarzinom; NSCLC) des rechten Oberlappens. Das PET-CT lieferte keinen Hinweis auf eine Fernmetastasierung, zeigte jedoch zusätzlich einen auffällig vermehrten Stoffwechsel und eine Inhomogenität der Muskulatur insbesondere im Schulter- und Hüftgürtelbereich, welches als radiologisches Korrelat zu einer Muskelbeteiligung im Rahmen der DM angesehen wurde. In der ergänzenden Tumorsuche mittels Gastroskopie und Coloskopie fanden sich keine Hinweise auf ein weiteres Malignom.
Wir leiteten initial eine Therapie mit Prednisolon (80 mg/d) und Azathioprin (100 mg/d) ein. Aufgrund des hohen Leidensdruck des Patienten entschlossen wir uns zusätzlich einen Zyklus hochdosierte intravenöse Immunglobuline (IVIG) zu geben (2 g/kg Körpergewicht [KG]). Unter dieser Therapie kam es zu einer schnellen Besserung des Allgemeinbefindens, der schmerzenden Erosionen, der Ulzera und der groben Kraft. Vier Wochen später wurde der Tumor mittels Oberlappenektomie rechts und Lymphadenektomie entfernt und im Verlauf von vier Monaten mit vier Zyklen Chemotherapie (Carboplatin, Vinorelbin) behandelt.
Eine erneute Vorstellung sechs Monate nach Erstdiagnose der DM zeigte einen rückläufigen Hautbefund mit abgeblassten Erythemen, verheilten Ulzerationen und einer residuellen Poikilodermie (Abbildung 3a–d). Laborserologisch waren die erhöhten CK-Werte im Verlauf gesunken (Abbildung 4). Die medikamentöse Therapie mit Azathioprin konnte beendet und das Prednisolon langsam ausgeschlichen werden.
Diskussion
In unserem Fallbericht zeigen wir einen seltenen, aber didaktisch wichtigen Fall einer bullösen DM als Paraneoplasie. Ein Malignom kann entweder bereits vor der Diagnose der DM bekannt sein, bei Erstmanifestation der DM im Rahmen der Untersuchungen festgestellt werden oder erst danach auftreten. Ungefähr 15 % der Patienten mit DM, besonders diejenigen über 40 Jahre, haben oder entwickeln im Verlauf ein Malignom [1–3]. In manchen Studien wird ein sechsfach erhöhtes Risiko einer Malignomentstehung angenommen. Das Risiko ist in den ersten drei Monaten nach Erstmanifestation am höchsten und bleibt bis zu fünf Jahre lang erhöht [3, 4]. Zu den häufigsten Tumoren bei einer paraneoplastischen DM gehören die soliden Tumoren der Lunge, des Ovars, des Magen- Darm-Trakts, des Pankreas sowie Non-Hodgkin-Lymphome [1, 4].
Die beschriebenen Hautveränderungen zeigen zum einen charakteristische Zeichen einer DM wie Erytheme im Nacken (shawl sign), im Dekolleté (V-sign), an den Hüften (holster sign), scheckige Hyperpigmentierung (Poikilodermie) und ein zentrofaziales Erythem, während andere typische Hauterscheinungen wie der weinerliche Gesichtsausdruck durch ein charakteristisches fliederfarbenes, periorbitales Ödem (heliotropes Erythem) und erythematöse, lichenoide, konfluierende Papeln über den Metacarpophalangeal- und Interphalangealgelenken (Gottron-Papeln) fehlen [2, 5]. Zudem weist der Patient ungewöhnlich seltene Hauterscheinungen einer vesikulobullösen oder erythroödematösen DM auf, die 1951 von Findlay das erste Mal beschrieben wurde und gegenwärtig zunehmend als paraneoplastisches Phänomen diskutiert wird [3, 6]. Diese vesikulobullöse Form kann auftreten in Form von kleineren Vesikeln und Erosionen, aber auch in Form von großen Bullae und findet sich oftmals am Stamm, den Extremitäten und im Gesicht [7–9], ähnlich wie bei unserem Patienten. Es lässt sich zu dem klinischen Erscheinen auch ein histologisches Korrelat finden.
Die vesikulobullösen Läsionen kommen durch ein Ödem in der oberen Dermis mit zum Teil reichlichen Muzinablagerungen zustande [6]. In der Literatur wird eine schlechtere Prognose und eine höhere Malignominzidenz bei Patienten mit vesikulobullösen Formen der DM beschrieben, welche mit einer höheren Morbidität und Mortalität einhergehen kann [3, 9–11]. In unserem Fall konnte dies bestätigt werden.
Es werden auch Myositis-spezifische Antikörper (MSA) diskutiert, die mit einer paraneoplastischen DM korrelieren. In unserem Fall beinhalten die abgenommenen Myositis-spezifischen Antikörper leider nicht den Antikörper Anti-TIF1-γ oder NXP2, die bei einer paraneoplastischen DM eine hohe Bedeutung zu haben scheinen [12, 13]. Zum damaligen Zeitpunkt (2014) waren TIF1-γ- und NXP2-Assays noch nicht breit verfügbar. Neben MSA wurden bei Patienten mit DM auch Myositis assoziierte Antikörper (MAA) beschrieben, welche von anderen Kollagenosen bekannt sind, wie anti-Ro-, -La-, -U1-RNP- und –Zentromer-Antikörper, um nur einige zu nennen. Bei paraneoplastischen Manifestationen der DM wird diskutiert, dass sowohl die antitumorale Immunantwort als auch das Freiwerden multipler zellulärer Antigene unter Chemotherapie zu Antigen- oder Epitop-Erweiterung führen kann [14, 15].
Als Goldstandard in der Therapie der DM werden systemische Glukokortikosteroide eingesetzt, oftmals in Kombination mit DMARDs (disease-modifying anti-rheumatic drugs) als zusätzlich immunsuppressive Medikation und zur Steroideinsparung. Am häufigsten werden Azathioprin oder Methotrexat gegeben [2, 4]. Bei schweren Formen der DM, wie einer paraneoplastischen DM, ist die Indikation zur Therapie mit IVIG gegeben und kann auch als First-Line-Behandlung
im Sinne einer adjuvanten Behandlung vertreten werden [16, 17].
In unserem Fall korrelierten die Hauterscheinungen mit dem Auftreten eines Malignoms und verschwanden oder waren rückläufig nach der Entfernung des Tumorgewebes. Dies zeigt zum einen als didaktisches Beispiel den Zusammenhang der Hauterscheinungen und der Paraneoplasie, und zum anderen, dass der hier beschriebene Fall einer bullösen DM wie schon in anderen Fallberichten wahrscheinlich in Zusammenhang mit einem paraneoplastischen Phänomen stehen kann.
Dies unterstreicht die Notwendigkeit einer akribischen Tumorsuche bei Patienten mit DM genauso wie den Umstand, dass die bullöse Form als Differenzialdiagnose in Abgrenzung zu anderen blasenbildenden Erkrankungen in Betracht gezogen werden kann [8, 13].
Interessenkonflikt
Keiner.
Literatur
1 Findlay AR, Goyal NA, Mozaffar T. An overview of polymyositis and dermatomyositis. Muscle Nerve 2015; 51: 638–56.
2 Volc-Platzer B. [Dermatomyositis-update]. Hautarzt 2015; 66: 604–10.
3 Zarrabi K, Choy T, Sweeney K et al. Paraneoplastic edematous dermatomyositis: A rare syndrome observed in a case of small cell lung cancer. Clin Pract 2017; 7: 982.
4 Mainetti C, Terziroli Beretta-Piccoli B, Selmi C. Cutaneous manifestations of dermatomyositis: a comprehensive review. Clin Rev Allergy Immunol 2017; 53: 337–56.
5 Schempp CM, Schauer F, Huhn CK et al. Skin inflammation associated with arthritis, synovitis and enthesitis. Part 2: rheumatoid arthritis, reactive arthritis, Reiter's syndrome, Lyme borreliosis, dermatomyositis and lupus erythematosus. J Dtsch
Dermatol Ges 2019; 17: 167–81.
6 Levy L, Layher H, McNiff JM et al. Dermatomyositis: histopathologic findings of parakeratosis and dermal edema revisited. J Cutan Pathol 2018; 45(4): 282–5.
7 Fujimoto M, Murakami T, Murata S et al. Acute onset vesiculo- bullous dermatomyositis associated with massive mucosal necrosis of the intestines. Clin Exp Dermatol 2002; 27: 718– 20.
8 McCollough ML, Cockerell CJ. Vesiculo-bullous dermatomyositis. Am J Dermatopathol 1998; 20: 170–4.
9 Tu J, McLean-Tooke A, Junckerstorff R. Increasing recognition of dermatomyositis with subcutaneous edema – is this a poorer prognostic marker? Dermatol Online J 2014; 20: 21244.
10 Kubo M, Sato S, Kitahara H et al. Vesicle formation in dermatomyositis associated with gynecologic malignancies. J Am Acad Dermatol 1996; 34: 391–4.
11 Zangrilli A, Papoutsaki M, Bianchi L et al. Bullous dermatomyositis: a marker of poor prognosis and aggressive internal malignancy? Acta Derm Venereol 2008; 88: 393–4.
12 Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med 2016; 280: 8–23.
13 Schiffmann ML, Warneke VS, Ehrchen J. Amyopathic dermatomyositis with anti-TIF1 gamma antibodies. J Dtsch Dermatol Ges 2018; 16: 76–78.
14 Hashimoto T, Muneta K, Watanabe K. Scleroderma/dermatomyositis overlap syndrome with anti-centromere and U1 RNP antibodies during chemotherapy for gastric cancer. J Dtsch Dermatol Ges 2017; 15: 561–2. Diagnosequiz 270 © 2020 Deutsche Dermatologische Gesellschaft (DDG). Published by John Wiley & Sons Ltd. | JDDG | 1610-0379/2020/1803
15 Vanderlugt CL, Miller SD. Epitope spreading in immunemediated diseases: implications for immunotherapy. Nat Rev Immunol 2002; 2: 85–95.
16 Enk A, Hadaschik E, Eming R et al. European Guidelines (S1) on the use of high-dose intravenous immunoglobulin in dermatology. J Dtsch Dermatol Ges 2017; 15: 228–41.
17 Hoffmann JHO, Enk AH. High-dose intravenous immunoglobulins for the treatment of dermatological autoimmune diseases. J Dtsch Dermatol Ges 2017; 15: 1211–26.
Die Diagnosequizze werden uns freundlicherweise zur Verfügung gestellt vom
"Journal der Deutschen Dermatologischen Gesellschaft" © Deutsche Dermatologische Gesellschaft